研究テーマ概要¶
English version is available here .
第一原理量子モンテカルロ法 (FP-QMC)¶
物質の化学的/物理的な性質を司る「電子」の量子力学的性質は、シュレーディンガー方程式を解くことにより、原理的には予測できる。しかし、特殊な例を除いて解析的な解は得られないため、通常は、大型計算機による第一原理計算により、数値的にその解を求める。現在、最も普及している第一原理計算は、波動関数理論 (Wavefunction theory)、もしくは、密度汎関数理論 (Density Functional Theory:DFT)に基づく方法であり、量子化学/材料科学/凝集系物理学分野において、数多くの成功を収めている。しかしながら、波動関数法においては、周期境界の扱いが難しい、電子数に対する計算コストのスケーリングが悪い等の問題点が、密度汎関数法においては、交換相関汎関数の選択によって大きく予見が変わる等の大きな問題点があり、より厳密な、次世代の電子状態計算開発が望まれてきた。
第一原理量子モンテカルロ法 (First-Principle Quantum Monte Carlo:FP-QMC)は、モンテカルロ積分を利用して多体シュレーディンガー方程式を数値的に解くことで物質の電子状態を解く方法であり、現在の電子状態計算手法の内で、最も厳密解に近い解が得られる手法である。

しかしながら、FP-QMCは、物性論、多体電子論、及び並列計算機に関する高度な知識と多くのノウハウを元に計算が行われるため、DFTのように誰もが気軽に利用できる計算手法ではない。現在は主に、FP-QMCの第一人者、SISSA (Italy)のSandro Sorella教授と協働し、TurboRVB の開発と応用を中心に研究を行っている。 TurboRVB は、イタリアのSISSAを中心に開発されている第一原理量子モンテカルロ法の計算コードである。
TurboRVB は、他の第一原理量子モンテカルロ法の計算コードと比べて、以下の点に特徴を有する。
- 多体波動関数として、Resonating Valence Bond (RVB)型のジャストロージェミナル/ジャストローパフィアン波動関数を実装しており、一般的なジャストロースレーター波動関数を採用する他の計算コードに比べて、電子相関をより正確に取り込んだ計算が可能。
- 変分量子モンテカルロ法における多体波動関数の振幅/節最適化を安定的に実行するStochastic ReconfigurationやLinear methodなどの優れた最適化アルゴリズムを実装。
- 格子離散化拡散量子モンテカルロ法 (Lattice regularized diffusion Monte Carlo)を実装しており、数値的に安定な第一原理拡散量子モンテカルロ計算が可能。
- トップダウン型自動微分法の実装により、多体波動関数の微分値を効率的に求めることができ、原子に働く力の計算/構造最適化/分子動力学なども実行可能。
これらの特徴を最大限に活かし、これまでのジャストロースレーター型で扱えなかった強相関系の電子状態計算や、第一原理量子モンテカルロ法レベルでの動的物性の計算等、に取り組んでいる。
密度汎関数理論 (DFT)に基づく第一原理計算の精度検証¶
近年の計算機、及び情報科学(AI関連)技術の急速な発展により、ビックデータに基いて新しい材料を探索、もしくは設計するという試み「Materials Informatics (MI)」に注目が集まっている。しかしながら、囲碁などのボードゲーム、製造現場における品質管理, Webデータの解析とは異なり、材料分野の最大の問題は、そもそも統一されたデータベースがなく、機械学習やデータマイニングをするためのビックデータがないことである。そこで、材料分野に関しては、理論計算で目的の物性が計算できる場合、理論計算によりその物性のデータベースを作成することがしばしば行われる。
密度汎関数理論 (Density Functional Theory)に基づく第一原理計算は、計算コストと精度の観点からそのようなデータベースを作成するための手法として最も現実的な手法である。しかしながら、DFT計算は全ての物性を正確に計算できるわけではなく、汎関数の不備、励起状態計算に関する手法の限界などから、実験値を再現できない時が多くある。したがって、ある物性値を計算するための理論が開発され、それが汎用ソフトに実装されていたとしても、その計算精度を幅広い化合物に対して確認することは、DFTの応用上非常に重要である。
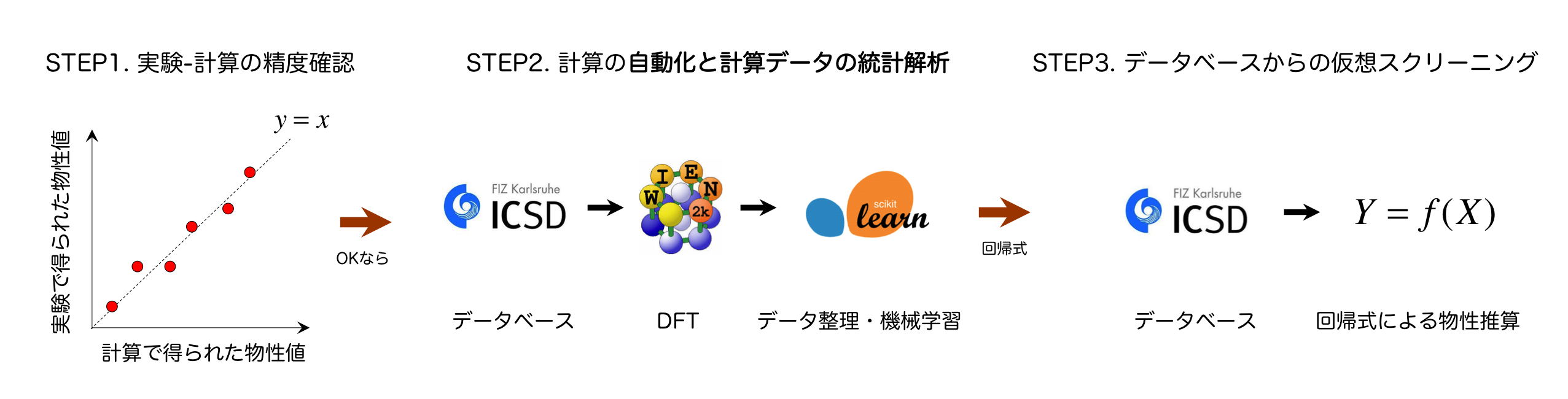
そこで、公知な実験データの収集、及びそのデータを利用したDFT計算の精度確認を行った。
計算材料科学を利用した材料物性機構の解明¶
近年の計算機、及び情報科学(AI関連)技術の急速な発展により、「Materials Informatics (MI)」に過度な注目が集まっているものの、元来、物理学における理論計算というのは、現実に起こる物理現象のメカニズムを解明するために利用されてきた経緯があり、そのような理論計算の使い方は、現在でも非常に重要である。理論計算の専門家の大きな役割として、実験系研究者と共同し、実験系研究者が新しく発見した材料の機構解明に役立つような理論計算を行っている。
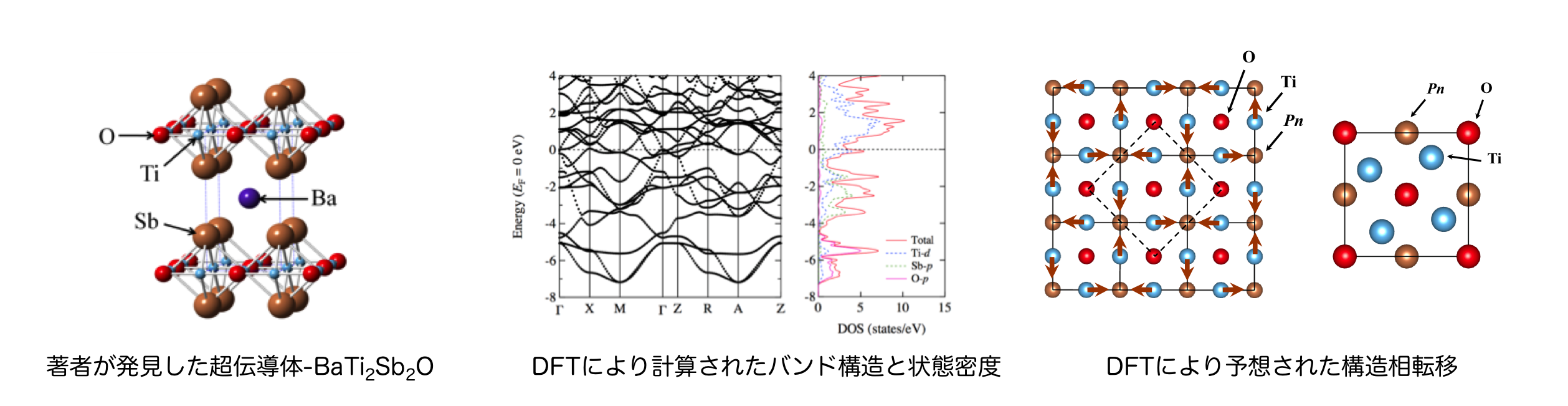
層状チタンニクタイド酸化物における新規超伝導体¶
多くの金属は極低温まで冷やすとある転移温度 (Tc)以下で電気抵抗が突然消失する.この現象は1911年にオランダの低温物理学者Kamerlingh OnnesによってHg (Tc = 4.2 K)で初めて発見され, 後に「超伝導 (superconductivity)」と名付けられた.新規超伝導体の探索は究極的にはTcが室温付近になる「室温超伝導」を目指して行われてきたが、2018年現在、未だにその目標は達成されておらず、その実現は人類の大きな夢となっている.
著者は今でこそ理論系研究者としてのキャリアを歩んでいるが、初めは実験系研究者として研究のキャリアをスタートさせた。その時に与えられた初めての研究テーマは新しい高温超伝導体の探索であった.そのテーマの過程で運良く(?)見つけた「層状チタンニクタイド酸化物」という化合物群に属する新奇超伝導体BaTi2Sb2O, 及びBaTi2Bi2Oは, 銅酸化物高温超伝導体や鉄砒素系高温超伝導体との類似性から、その超伝導機構や、超伝導に関連すると思われる低温での構造相転移機構に大きな注目が集まり、2012年の発見から6年が経過した今でも精力的に研究が続けられている.特に低温の構造相転移については、主に回折実験から、低温での構造を決定する試み、及び構造相転移機構の議論が多く成されたが決定打にかけていた。そこで、計算側からその構造の決定、及び相転移機構の議論を行うべく、第一原理計算を本物質群に適用し,層状チタンニクタイド酸化物の物性機構の解明を試みた.
実験系グループとの共同研究¶
自身が実験系研究者であった経験を活かし、積極的に実験系研究グループと共同研究を実施している。 具体的な依頼があった場合には、それら物質の電子状態計算に関してアドバイスを行う、 もしくは自ら手を動かして計算を行っている。